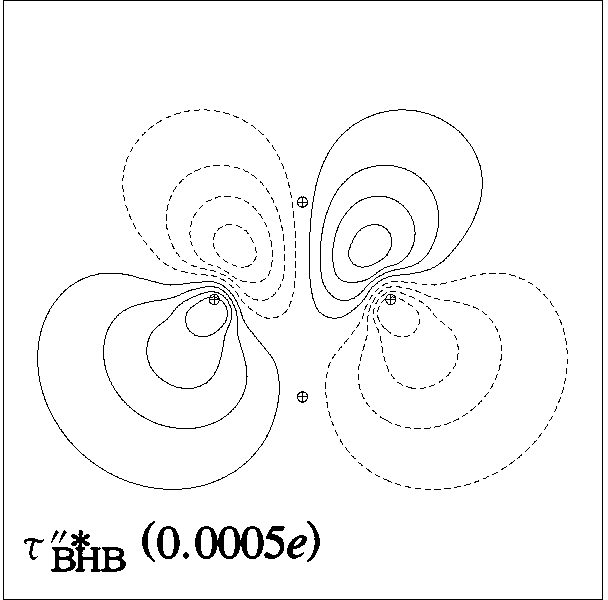Natural Orbitals (NOs) are the unique orbitals chosen by the
wavefunction itself as optimal for its own description. Mathematically,
the NOs {Θi} of a
wavefunction Ψ can be defined [1] as the
eigenorbitals of
the first-order reduced density operator Γ,
(1) ΓΘk = pkΘk (k = 1,2,...)
In this equation, the eigenvalue pk represents
the population (occupancy) of the eigenfunction
Θk for the molecular
electron density operator Γ of Ψ. The density operator is
merely the 1-electron
"projection" of the full N-electron probability distribution
(given by the square of the wavefunction |Ψ|2) for answering
questions about 1-electron subsystems
of the total wavefunction Ψ. Thus, Ψ is the only quantity
that enters into the definition of the NOs,
and these orbitals are truly Ψ's "own" (eigen) orbitals,
intrinsic ("natural") to description of the electron density
and other single-electron properties of Ψ. As for any
Hermitian eigenvalue problem, the NOs form a complete
orthonormal set.
Alternatively, we can characterize
{Θk}
as maximum occupancy orbitals. The electronic occupancy pφ
of any normalized "trial orbital" φ can be evaluated as
the expectation value of the density operator, viz.,
(2) pφ = <φ|Γ|φ>
Variational
maximization of pφ for successive orthonormal
trial orbitals
(3a) max<φ|Γ|φ> = p1 (best
φ is Θ1)
(3b) max<φ'|Γ|φ'> =
p2 (best φ' orthogonal to Θ1 is Θ2)
(3c) max<φ''|Γ|φ''>
= p3 (best
φ'' orthogonal to Θ1 and Θ2 is Θ3), etc.
leads to optimal populations pk
and orbitals Θk
that are equivalent to those
in Eq. (1),
as follows from general min/max properties of
eigenvalue equations. The Pauli exclusion principle
insures that these occupancies satisfy 0 ≤ pk ≤ 2.
Note that the wavefunction Ψ is commonly described
with the help of chosen "basis orbitals" {χk}
(such as those of a 6-311++G** basis set, as employed
in numerical work described below). However, for
given Ψ the solutions of Eq. (1) are in principle
independent of the chosen basis orbitals, whether
of Slater, Gaussian, or plane wave type. Indeed, the
NOs and associated orbital-based concepts remain rigorously defined by Eq. (1)
even if Ψ is specified only in
interparticle (Hylleraas or James-Coolidge type
rij)
coordinates, without reference to any
orbital basis set whatsoever. While it is often numerically
convenient to employ basis orbitals (orthogonal or non-orthogonal)
to solve eigenvalue problems such as Eq. (1), it is important
to realize that the eigenorbitals Θk are
in principle independent of the particular
basis orbitals chosen. Natural orbitals Θk
are intrinsic and unique to Ψ, whereas basis orbitals
χk
are non-unique "fitting" functions, chosen merely for numerical
convenience.
What are "Natural Atomic Orbitals" (NAOs)?
Natural Atomic Orbitals (NAOs) {Θk(A)} are localized 1-center
orbitals that can be described as the effective "natural orbitals of atom A"
in the molecular environment. We shall first summarize some qualitative
features that distinguish NAOs
from other types of "atomic orbitals" before presenting formal mathematical
definitions and details of the numerical algorithms
for determining these orbitals in the NBO program.
The NAOs incorporate two important physical
effects that distinguish them from
isolated-atom natural orbitals as well as from standard
basis orbitals:
(i) The spatial diffuseness of NAOs is optimized for the effective atomic
charge in the molecular environment (i.e., more contracted if A is
somewhat cationic; more diffuse if A is somewhat anionic). NAOs
therefore automatically incorporate the important "breathing" responses
to local charge shifts that usually require variational contributions
from multiple basis functions of variable range
(double zeta, triple zeta, or higher) to describe accurately.
(ii) The outer fringes of NAOs incorporate the important nodal
features due to steric (Pauli) confinement in the molecular environment
(i.e., increasing oscillatory features and higher kinetic energy
as neighboring NAOs begin to interpenetrate, preserving the
interatomic orthogonality required by the Pauli exclusion
principle). The valence NAOs of atom A therefore properly incorporate
both the inner nodes that preserve orthogonality to its own
atomic core as well as the outer nodes that preserve
orthogonality to filled orbitals on other atoms B. Both features
are necessary for realistic steric properties
in the molecular environment (i.e., proper Fermi-Dirac
anticommutators of the associated
second-quantized NAO field operators [2]),
but both are commonly ignored in standard basis orbitals.
A distinguishing hallmark of NAOs is their strict preservation
of mutual orthogonality, as mathematically required for eigenfunctions
of any physical Hermitian operator. Each
NAO therefore maintains intraatomic orthogonality
to remaining NAOs on the same
atom as well as interatomic orthogonality to those on other atoms, viz.,
(4) <Θj(A)|Θk(B)> = δj,kδA,B
However, the NAO numerical algorithms (see below) allow removal of interatomic
orthogonality to give the associated "pre-orthogonal NAOs" (PNAOs),
denoted {pΘk(A)}. Except for omission of long-range orthogonality
tails (and the accompanying steric pressure of surrounding atoms),
the PNAOs and NAOs are identical. PNAOs therefore
preserve the necessary radial and angular nodal features
to remain orthogonal within
each atom, but they overlap the PNAOs on other atoms, viz.,
(5a) <pΘj(A)|pΘk(A)> = δj,k
(5b) <pΘj(A)|pΘk(B)> ≠ 0 (A ≠ B)
PNAOs thus
exhibit the idealized spherical symmetries of isolated atoms,
but remain optimally adapted in size for the molecular
environment. Moreover, these orbitals exhibit the
interatomic orbital overlap that underlies
qualitative concepts of chemical bonding. PNAOs are therefore
the preferred choice as "textbook" atomic orbitals, providing
vivid imagery to illustrate
the principle of maximum overlap.
The PNAO overlaps
<pΘj(A)|pΘk(B)>
also allow one to visually estimate the strength of actual NAO
interaction energies (Fock or Kohn-Sham
matrix elements
<Θj(A)|F|Θk(B)>) by means of
Mulliken-type approximations of the form
(6) <Θj(A)|F|Θk(B)> =
−k <pΘj(A)|pΘk(B)>
where k is a proportionality constant of order unity. Thus,
the use of NAOs preserves the full mathematical rigor of
a physical (Hermitian, Pauli-preserving) Schrödinger-type
eigenvalue problem for the atomic subsystem in the molecular environment,
whereas the PNAOs allow a corresponding qualitative description in terms
of familiar overlap-based bonding concepts.
The NAOs are automatically ordered in importance by
occupancy. Consistent with chemical intuition, only the
core and valence-shell NAOs are found to have significant
occupancies, compared to the extra-valence Rydberg-type NAOs
that complete the span of the basis. The effective dimensionality
of the NAO space is therefore reduced to that of the formal
"natural minimal basis" (NMB), spanning core and valence-shell
NAOs only, whereas the residual "natural Rydberg basis" (NRB)
of extra-valence NAOs plays practically no significant role in NBO
analysis. This condensation of occupancy into the much
smaller set of NMB orbitals (allowing the large residual NRB set
from the original basis to be effectively ignored)
is one of most dramatic and characteristic
simplifying features of "natural" analysis.
Let us now describe some deeper aspects of the numerical
algorithms by which 1-center NAOs (and PNAOs) are obtained, and the
relationship to the multi-center NO definitions (1)-(3).
The NAO algorithm is usually assumed to start from
a standard basis set of atom-centered orbitals {χj},
each centered at the position of
nucleus A. But suppose that the chosen basis orbitals
are of more general form, such as plane waves {wk}. In this
case one may begin by calculating a wavefunction and density operator
for each isolated atom A (each centered at its proper position
in the molecule). From such calculations one obtains corresponding
free-atom NOs χj(A) (ordered by occupancy) for each atom
in the plane wave basis
(7) χj(A) = Σkcjk(A)wk
From the total number of such free-atom NOs, one can select the
leading χj(A)'s from each atom to prepare a composite
atom-centered basis {χj(A), χj'(B),...} = {χj} that
is linearly independent and spans the same dimensionality as the
original basis {wk}. The transformed single-center basis
orbitals {χj} therefore allow the NAO algorithm to
begin in the usual way. [Because atom-centered basis orbitals
are the near-universal choice in standard electronic structure
packages, the current NBO program makes no provision
for the hypothetical initial basis transformation (7).]
As typically obtained from the host electronic structure
program or the transformation (7), the starting atomic basis orbitals
{χj} are overlapping between nuclear
centers A,B,..., and the sum of their occupancies (2) exceeds
the actual total number of electrons N by nonorthogonal
overcounting. The key step is therefore to replace these
overlapping χj's by corresponding basis orbitals oχj that
are orthogonal between nuclear centers, viz.,
(8) <oχj(A)|oχj'(B)> = Sj,j'δA,B
The operator TOWSO
that performs the symbolic transformation to orthogonalized
basis orbitals {oχj}
(9) TOWSO{χj} = {oχj}
may be described as occupancy-weighted symmetric orthogonalization
(OWSO). The TOWSO transformation
can be shown (by the general Carlson-Keller theorem [3])
to variationally maximize the
"resemblance" between initial χj's and final oχj's (or
more mathematically, minimize their root-mean-square deviation) in
a population-weighted sense, i.e.,
(10) Σkpk ∫|χk − oχk|2 dτ
= minimum
where pk = <χk|Γ|χk> is the population
of χk. Initial basis orbitals of high occupancy are therefore highly
preserved by the OWSO transformation (10), whereas those
of low or negligible occupancy (corresponding, e.g., to diffuse
Rydberg-type orbitals, achieving "occupancy" only by
overlapping filled orbitals on other centers) are allowed to distort as
necessary to achieve overall minimization. The OWSO transformation
is intrinsically stable toward basis set extensions
and achieves the optimal separation of atomic subspaces
in a maximally democratic manner. It is then straightforward
to define the subsystem density operator Γ(A) for atom A (in matrix
representation, the matrix of Γ in the subspace of oχk's
for that atom). In analogy to Eq. (1), the NAOs {Θk(A)} of atom A
are defined as eigenorbitals of Γ(A)
(11) Γ(A)Θk(A) = pk(A)Θk(A) (k = 1,2,...)
with corresponding eigenvalues (populations)
pk(A) = <Θk(A)|Γ(A)|Θk(A)>
= <Θk(A)|Γ|Θk(A)>. Alternatively, the Θk(A)'s can be
defined as 1-c "maximum occupancy" orbitals by operations
analogous to (3) for the 1-c operator Γ(A). The full NAO algorithm [4]
involves other technicalities, but its essentials
are captured in Eqs. (8)-(11).
The NAOs also underlie "Natural Population Analysis" (NPA),
which has widely supplanted "Mulliken Population Analysis" [5]
for ab initio description of atomic charge distributions. The
NAO populations pk(A) sum properly to the total number of
electrons N and lead unambiguously to the corresponding
net natural atomic chargeQA
(12) QA = ZA − Σkpk(A)
on each atom A (nuclear charge ZA). The
natural populations automatically satisfy physical positivity and
Pauli constraints (0 ≤ pk(A) ≤ 2 for closed shells), and the
stability of the OWSO-based NAOs automatically
insures numerical stability of NPA populations and atomic charges
with respect to basis set extensions. [This contrasts sharply
with Mulliken populations, which are often found to exhibit unphysical
negative or Pauli-violating values and numerical instabilities that
tend to increase as the basis set is
extended; indeed, as shown by Ruedenberg [6], Mulliken
populations can actually exhibit any value between ±∞ in the limit
of a complete basis!]
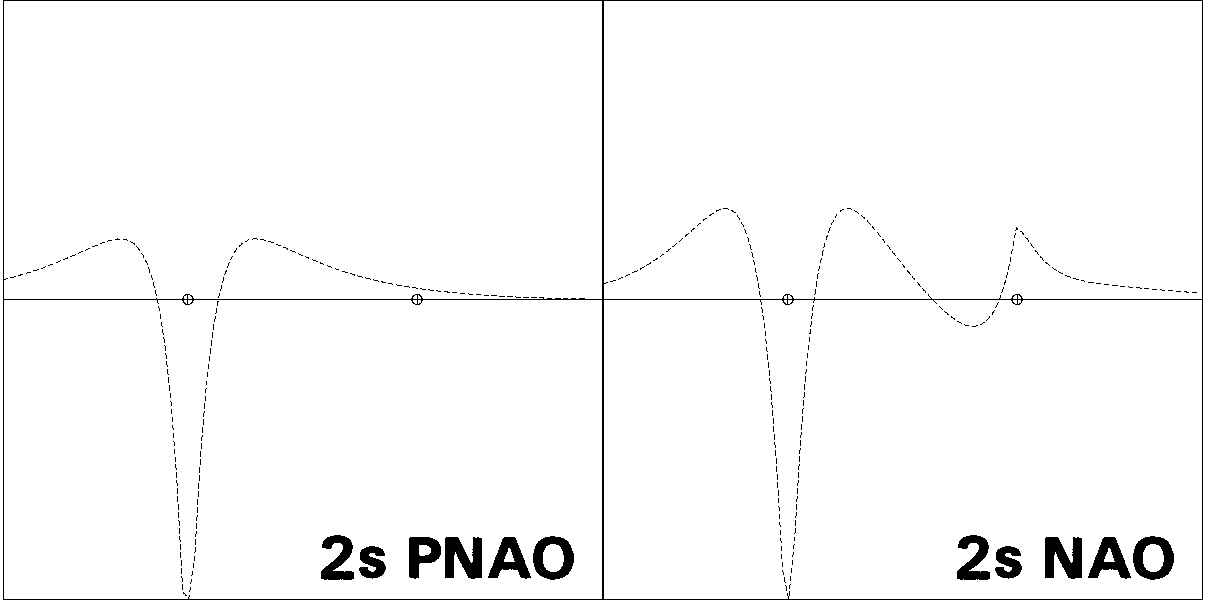
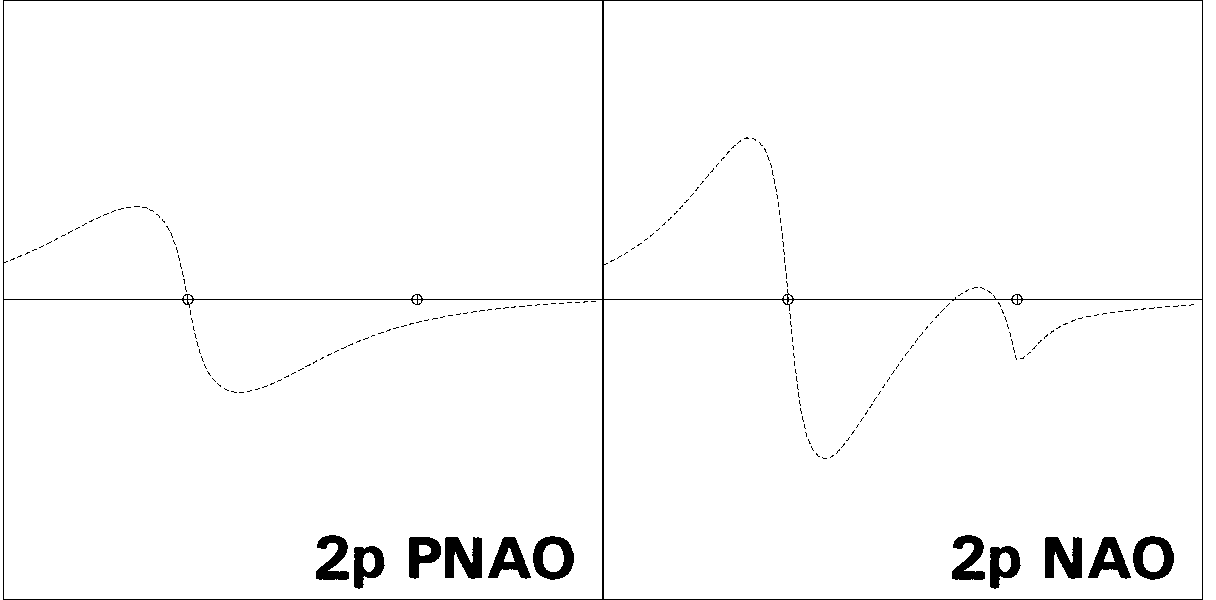
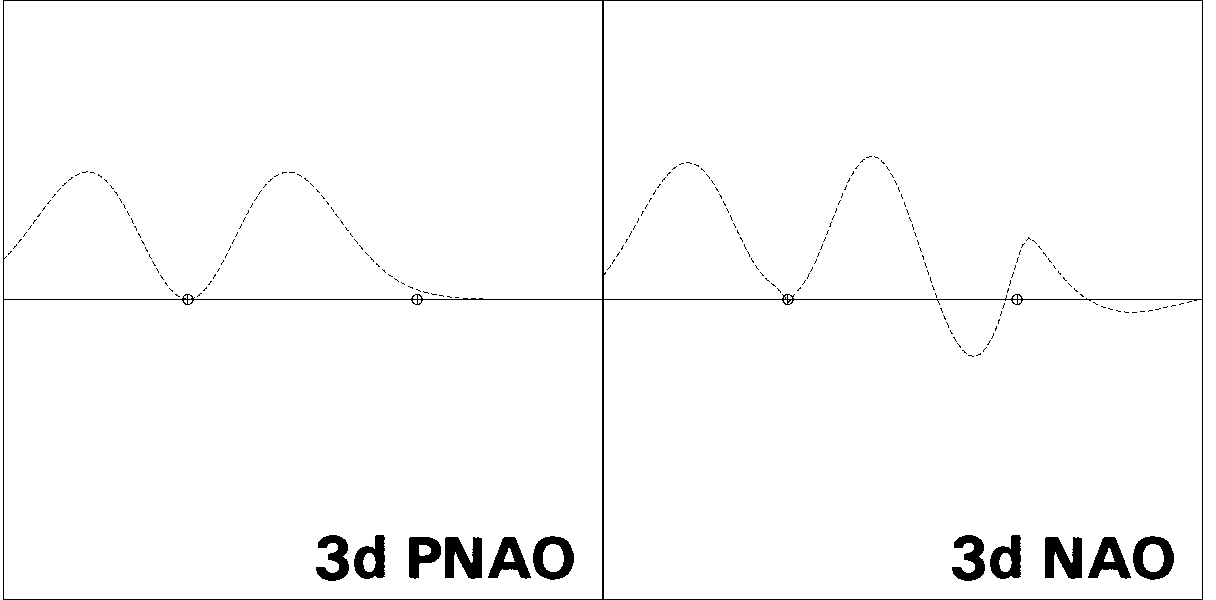
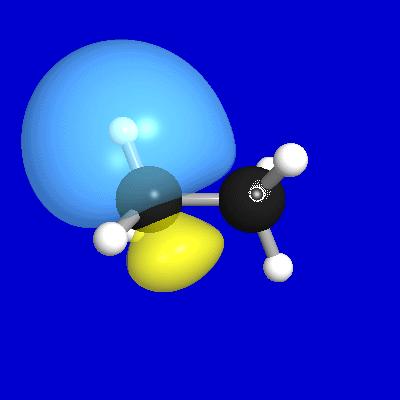
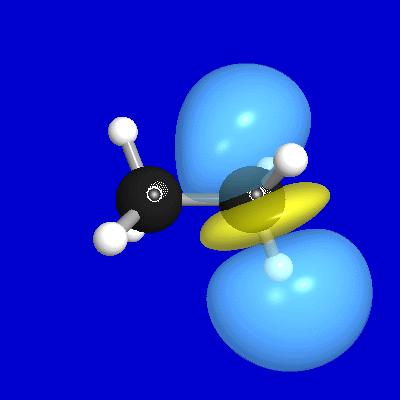
Figures 1-3 illustrate some 1-D, 2-D, and 3-D comparisons of
valence and Rydberg NAOs and PNAOs for
one of the carbon atoms of
ethane (B3LYP/6-311++G** level). The oscillations between positive
and negative phase, shown with respect to the horizontal "zero" line
in Fig. 1, are represented in Fig. 2 by solid vs. dashed
contour lines, or in Fig. 3 by blue vs. yellow lobe surfaces.
Figure 1: PNAO/NAO Radial Profiles
Figure 2: PNAO/NAO Contour Diagrams
Figure 3: PNAO/NAO 3-D Surfaces
Each figure displays
the valence 2s, 2pz and Rydberg 3dz2
orbitals (with z direction lying along the internuclear CC axis,
and the nuclear positions marked with bullets), comparing
the PNAO (left) with the NAO (right). Fig. 1
shows the PNAO/NAO comparisons
in 1-D "profile" form, for orbital amplitude plotted along the CC axis.
The PNAO profiles are seen to exhibit the
expected nodal patterns of pure hydrogen-like 2s, 2p,
and 3d atomic orbitals in free space, whereas the corresponding NAO
profiles show a pronounced secondary nodal feature near the
adjacent C nucleus, maintaining orthogonality to the core electrons
occupying this region. In each case one can also see
the steric crowding of each NAO, leading to
contraction of outer lobes from the C(2) region and increased "peaking"
near the C(1) nucleus. Fig. 2 shows the corresponding
comparisons as 2-D contour
diagrams (in a plane that also contains two off-axis H
nuclei). Again, the PNAOs show the expected angular symmetries
of free-space s, p, d orbitals, whereas the NAOs
show the distinct compressional distortions due to the asymmetric molecular
environment, including nodal "tails" near adjacent
nuclei. Finally, Fig. 3 presents a similar comparison in rendered 3-D
surface images (showing
only the shape of the outermost contour in Fig. 2). Both Figs. 2, 3
emphasize that
the actual symmetry of a "2s" NAO
is considerably lower than the labelling might seem to imply
(the symmetry label applying strictly only to the parent PNAO). Nevertheless,
this asymmetrically distorted orbital is indeed playing the role of
the "effective" 2s orbital
in the molecular environment, and its calculated energy
will better match the inferred experimental value (e.g., from
photoelectron spectroscopy) than will an idealized free-atom orbital (e.g.,
the 2s PNAO).
Indeed, the energy difference
between PNAOs and NAOs underlies the STERIC keyword evaluation
of interatomic steric effects [7] in the NBO analysis
program. The numerical success of this analysis in evaluating
realistic rare-gas interaction potentials [7a]
and atomic van der Waals radii [7b] provides further evidence
that important effects of steric repulsion
are properly incorporated in the NAO energetics.
The occupancies ("natural populations") of
2s (1.0909e), 2p (1.0738e), and 3d (0.0011e) NAOs
also conform well to the
expected partial filling of the n = 2 valence shell
and essential vacancy of the n = 3 and higher Rydberg shells.
(Note that the occupancies 1.2006e of 2px and
2py NAOs differ slightly from that of 2pz quoted above,
reflecting the asymmetries of the C atom in the actual
ethane environment.)
In summary, both the free-atom-like
PNAOs and sterically distorted NAOs
serve complementary purposes, bringing qualitative
imagery (PNAOs) and quantitative energetic detail (NAOs) to the concept
of "atomic orbitals in the molecular environment." The PNAOs make
close contact
with the concept of the "effective minimal basis of AOs" that
underlies qualitative bonding theories, whereas the NAOs
provide the high-accuracy representation of complex electronic properties
in a highly compact and interpretable form. (The latter property is
surprising to those who have only attempted "chemical interpretation"
with standard basis AOs.)
What are "Natural Bond Orbitals" (NBOs)?
Natural Bond Orbitals (NBOs) are localized
few-center orbitals ("few" meaning typically 1 or 2,
but occasionally more) that describe the Lewis-like molecular
bonding pattern of electron pairs (or of individual electrons
in the open-shell case) in optimally compact form. More
precisely, NBOs are an orthonormal set of localized "maximum occupancy"
orbitals whose leading N/2 members (or N members in the
open-shell case) give the most accurate possible
Lewis-like description of the total N-electron density.
Neither
the form of the bonding hybrids nor the locations of localized
bonds and lone pairs are pre-determined. Rather, the
NBO program searches over all possible ways of drawing the
bonds and lone pairs for the variationally optimal bonding pattern
that places maximum occupancy (highest percentage of the total
electron density) in the leading N/2 "Lewis-type"
NBOs (typically >99.9%
for common organic molecules). The Lewis-type NBOs determine
the localized Natural Lewis Structure (NLS) representation of
the wavefunction, while the remaining "non-Lewis"-type NBOs
complete the span of the basis and
describe the residual "delocalization effects" (i.e., departures
from a single localized Lewis structure). Thus, NBOs provide
a valence bond-type description of the wavefunction,
closely linked to classical Lewis structure concepts. As in
the NAO case, the only input to the NBO algorithms is the
molecular wavefunction Ψ (through its first-order reduced
density operator Γ), so the numerically determined Lewis structure
representation is "natural" to Ψ itself.
In contrast to other "flavors" of NOs, MOs,
and LMOs, the NBOs enjoy a unique association with Ψ,
even in the limit of single-determinant Hartree-Fock or Kohn-Sham DFT
description. As is well known (cf. Ref. [12] and discussion below),
in closed-shell HF/DFT theory
the NOs, MOs, and LMOs are all doubly
occupied and maximally degenerate, hence mutually equivalent and
essentially indeterminate with respect to arbitrary unitary transformations
of no physical consequence. In
contrast, the occupancies of NBOs are generally non-degenerate,
and their occupancy variations reflect
physically important resonance delocalization corrections
to the idealized Lewis structure picture. Except for highly unusual
"accidental" degeneracies, each NBO is therefore uniquely determined
by its defining local-eigenorbital property, whether Ψ is correlated
or uncorrelated. The observed insensitivity of NBOs with respect
to variations of theoretical basis or method reflects their unique
association with the limiting Ψ, independent of approximation strategy.
The NBOs are composed of Natural Hybrid Orbitals (NHOs) {hA},
each being an optimized linear combination of NAOs on the given center
(13) hA = ΣkakΘk(A)
(The pre-orthogonal PNHOs are defined by an expansion with
identical ak's, but in terms of PNAOs rather than NAOs.)
Like the NAOs, the NHOs form a complete orthonormal set that spans
the full basis space. Core NBOs
(labelled "CR" in NBO output) are typically of nearly pure NAO
character. The 1-center
"lone pair" (nonbonding) NBOs nA (labelled "LP"
in NBO output) are each composed of a single
normalized NHO
(14) nA = hA
whereas the 2-center "bond" NBOs ΩAB (labelled "BD" in
program output) are normalized linear
combinations of two bonding NHOs hA, hB, corresponding to the
classical bond-orbital formulation of Mulliken and Lennard-Jones,
(15) ΩAB = aAhA + aBhB
with "polarization coefficients" aA, aB satisfying
aA2 + aB2 = 1. Depending on the values of these
coefficients, a bond NBO may range between covalent (aA = aB)
and ionic (aA >> aB) limits. However,
no sharp distinction can be drawn between a "2-center" ΩAB of highly polar
form (aA >> aB) and a "1-center" nA (aA = 1, aB = 0). To
conform to common chemical usage, the NBO program identifies
a highly polar ΩAB as a "lone pair" nA whenever 95%
or more of the electron density is on a single center
(aA2 ≥ 0.95).
Each in-phase NBO (15) of valence hybrids hA, hB
must be orthogonally complemented by the corresponding out-of-phase
antibond NBO Ω*AB (labelled "BD*" in program output)
(16) Ω*AB = aBhA − aAhB
Valence antibonds Ω*AB are
of non-Lewis type, having no role in the ground-state NLS description,
and the very term "antibond" may seem to suggest antagonism to
chemical stability and association. However, the antibonds derive from the
same unfilled valence hybrids that give rise to the bonding
NBOs, and the Ω*AB's therefore represent unused valence
shell capacity of the constituent atoms, unsaturated by covalent
bond formation. The Ω*AB's are typically found to be
the most important non-Lewis "acceptor" orbitals,
contributing to resonance stabilization,
intermolecular H-bonding, and other forms of supramolecular
donor-acceptor aggregation. Knowledge of the shapes and energies
of available Ω*AB antibond NBOs is the key to understanding
a host of important "non-covalent" and "delocalization" phenomena
that lie beyond the idealized Lewis structure picture.
Finally, the valence non-Lewis NBOs {Ω*AB} are complemented
by a set of "Rydberg-type" 1-center
NBOs rA (labelled "RY*" in program output)
that complete the span of the NBO basis. Like the extra-valence NAOs
from which they derive, these NBOs typically have negligible occupancy
and can be ignored for chemical purposes. The effective dimensionality
of significantly occupied NBOs is therefore reduced to that of the NMB
set of NAOs, in accordance with chemical intuition.
Table I summarizes characteristics of the common NBO types,
showing the number of centers, quantum shell, Lewis(L)/non-Lewis(NL)
donor-acceptor type, and NBO program output label:
Table I
NBO Type
centers
shell
L/NL
label
core cA
1-c
core
L
CR
nonbonded (lone pair) nA
1-c
valence
L
LP
bond ΩAB
2-c
valence
L
BD
antibond Ω*AB
2-c
valence
NL
BD*
Rydberg rA
1-c
Rydberg
NL
RY
unfilled nonbonded nA*
1-c
valence
NL
LV
3-c bond τABC
3-c
valence
L
3C
3-c antibond τ*ABC
3-c
valence
NL
3C*
The lower part of Table I also lists some less common NBO types. These
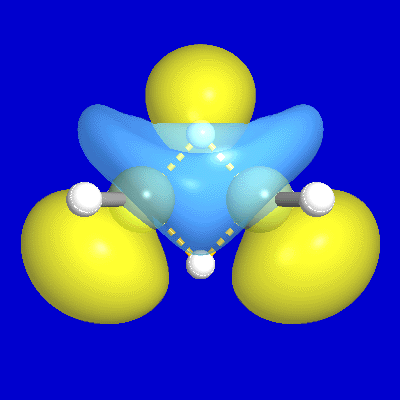
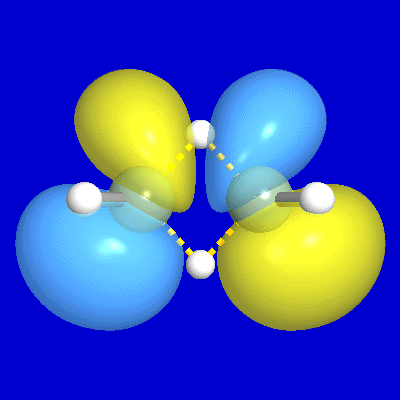
include unfilled valence nonbonding orbitals of "lone vacancy" (LV) type, as exemplified
by the unfilled valence 2pB orbital of boron in
BH3, as well as the 3-center
bonds (τABC) and antibonds (τ*ABC; actually, two
for each τABC) of B2H6
and related hypovalent species, as illustrated
in Figure 4. The search for 3-c bonds formerly required the 3CBOND keyword to be activated, but is now
included in default Lewis structure search. 3-c bonds seldom occur in ordinary
organic species, but for diborane
and other semi-metallic species such bonds qualitatively
improve the NLS description (e.g., raising the Lewis density
of the NLS wavefunction from 86.1% to 99.6% of the total density
in B3LYP/6-311++G** diborane). This
provides direct numerical evidence that a qualitatively new type
of localized bonding is needed to describe these species,
in accordance with the
prescient picture of Longuet-Higgins, Lipscomb, and others.
Figure 4: B2H6 3-c Bond, Antibond NBOs
Still
other possibilities beyond those listed in Table I may occur in excited
states. For example,
electronic excitation may leave a bonding ΩAB
vacant (hence, a non-Lewis "acceptor" orbital) while an antibonding Ω*AB
or Rydberg orbital rA is filled (hence, becoming a formal
Lewis-type "donor" orbital) in the best localized description. Such
exotic NBO types are now handled more consistently in NBO 7.9 than
in previous NBO versions.
A generic 2-c bond NBO ΩAB can often
be further classified according to local diatomic symmetry as
sigma (σAB) or pi (πAB) type. We emphasize that
the NBO procedure imposes no constraints in this regard, and indeed
the optimal forms of double bonds are occasionally found to exhibit
some degree of "banana bond" character. Similarly, there is no
intrinsic bias for whether two lone pairs have symmetrical ("rabbit ears")
or unsymmetrical (sigma-type vs. pi-type) character, but the latter
is typically found to be distinctly superior in the maximum occupancy
sense. The tendency of variationally optimized NBOs to resemble
"textbook-like" sigma and pi bonds is testimony to the deep intuition
of pioneer bonding theorists who were able to conceive these
idealized forms without benefit of accurate polyatomic wavefunctions.
Let us now briefly sketch some features of the NBO
program algorithm. In the methodical search for high-occupancy
1-c, 2-c orbitals throughout the molecule, a key step
is formation of the 2-c density operator aΓ(AB) for each
candidate atom pair A, B. This operator is the projection
of Γ on the complete basis of NAOs for the two atoms (including
off-diagonal elements connecting the 1-c Γ(A) operators discussed
previously). High-occupancy eigenorbitals aΩAB of this
operator
are partitioned into their normalized hybrid contributions
from each atom [cf. (15)]. For specificity, we denote by ahA(B)
the hybrid contribution on atom A directed
toward atom B, and ahB(A) as that on B directed
back toward A. In the search for candidate high-occupancy 2-c
orbitals to all other atoms, we acquire a provisional set
of hybrids ahA(B), ahA(B'), ahA(B''),..., each drawn freely
from the space of available NAOs without regard to possible
conflicts with bonds to other centers (possible
nonorthogonal overcounting). The provisional overlapping
ahA(B)'s are therefore
transformed by symmetric orthogonalization to final
orthonormal hA(B)'s (the NHOs), preserving maximum resemblance to
the parent ahA(B)'s in the mean-squared sense while restoring
inter-hybrid orthogonality,
(18a) ΣB ∫|ahA(B) − hA(B)|2 dτ =
minimum
(18b) <hA(B)|hA(B')> = 0, all B, B'
From these NHOs one can finally form
the projected 2-c density operator hΓ(AB) (projected onto
the two specific NHOs for bonding A to B) whose eigenfunctions
and eigenvalues (one for ΩAB, the other for Ω*AB) give the
final NBOs and occupancies, viz.,
(19) hΓ(AB) ΩAB = pAB ΩAB
Parallel to the 1-center NAOs (11), the NBOs (19)
can therefore be described as "local (2-c) eigenvectors of the
density operator," preserving the maximum-occupancy, orthonormality,
and completeness properties of many-center NOs (1).
In practice, the numerical
"pair" threshold pthresh of (17) is automatically scanned
through a grid of values, with the
optimal (maximum occupancy) Lewis structure being recorded for each
threshold value. The program searches
successively for 1-c, 2-c, 3-c pairs, removing the density contributions
of 1-c pairs (by rigorous orthogonal projection;
previously by "depletion" approximation) before beginning the search for 2-c pairs,
and so forth. The program finally reports the NBOs
of highest Lewis occupancy (or lowest non-Lewis occupancy)
in the overall search sequence. Consult
the original papers
[8]-[10], the NBO program manual [11], and detailed commentary within
the Fortran source code for further algorithmic details.
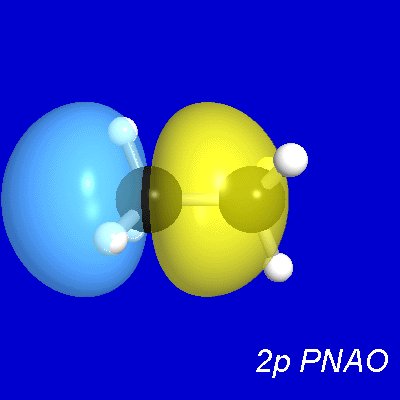
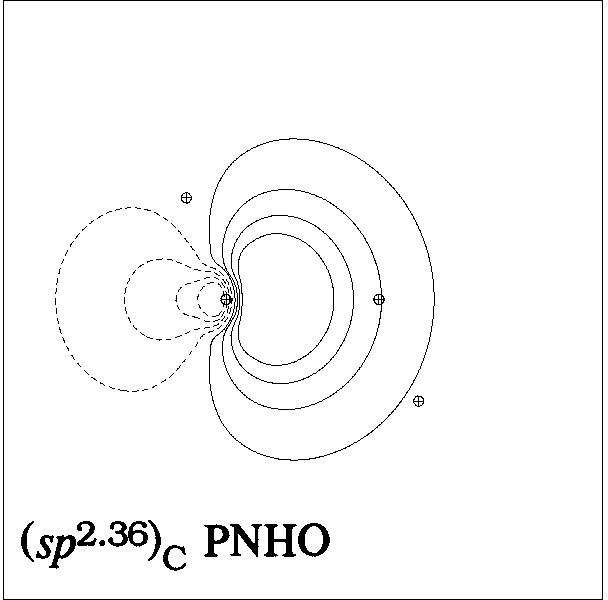
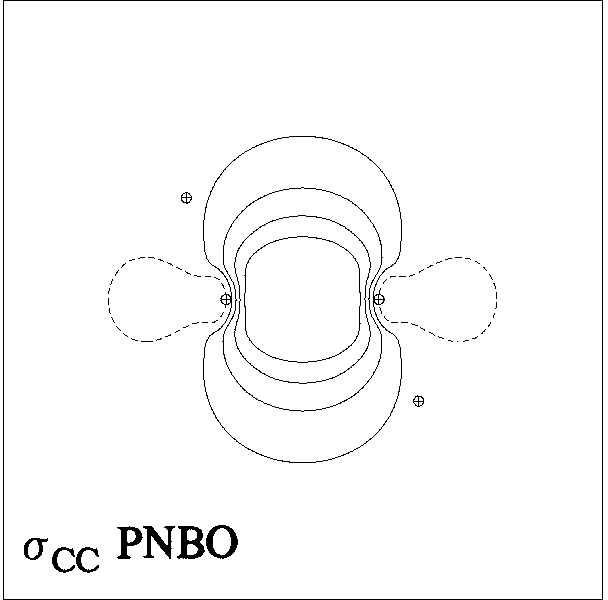
Figure 5 presents representative
contour and surface diagrams for the PNHOs, PNBO, and NBO of
the σCC bond of ethane, which can be expressed as [cf. (15)]
(20) σCC' = 0.707(sp2.36)C + 0.707(sp2.36)C'
Figure 6 shows similar contour and surface diagrams for
the overlapping hybrids and bond (σCH) and antibond (σ*CH)
(P)NBOs of one of the CH bonds, where the in-phase σCH
can be expressed as
(21) σCH = 0.773(sp3.25)C + 0.635(s)H
The carbon NHOs correspond reasonably
closely to the expected "sp3" hybridization of a Pauling-like
picture,
and the hydrogen NHO is essentially the expected pure 1s NAO. As shown
in the upper panels of Figs. 5, 6, the PNHOs exhibit the overlapping
angular shapes that are expected to lead to strong covalent bonding
in accordance with the "principle of maximum overlap".
Figure 5: CC Bond NHOs and (P)NBO
Figure 6: CH Hybrids and L/NL NBOs
What are "Natural Localized Molecular Orbitals" (NLMOs)?
NLMOs {ωi}
can be described as semi-localized alternatives to the ordinary
("canonical") CMOs for representing the electron pairs of
MO-type wavefunctions. Each NLMO ωi closely resembles
a "parent" NBO Ωi (strictly localized), but captures the
associated delocalizations needed to describe the density
of a full electron pair, thereby becoming a valid (non-canonical)
solution of the Hartree-Fock (or DFT-type)
SCF equations. Compared to CMOs, the NLMOs are
free from the superfluous constraints of
overall symmetry adaptation. NLMOs therefore adopt the
characteristic bonding pattern of a localized Lewis
structure, averting the symmetry-imposed mixings
(even between remote groups, beyond empirical van der Waals separation)
that limit transferability and interpretability of CMOs.
Consider, for example,
a system consisting of two
symmetry-equivalent CH bonds, one (σCH(↑)) at the North Pole
and one (σCH(↓)) at the South Pole. In the canonical MO formalism,
the electron pairs of this
system must be described
by "completely delocalized" CMOs φ1, φ2
of symmetric and antisymmetric form, viz.,
(22a) φ1 = 2−1/2[σCH(↑) + σCH(↓)]
(22b) φ2 = 2−1/2[σCH(↑) − σCH(↓)]
The CMO determinantal wavefunction
(23) ΨCMO = det|...(φ1)2(φ2)2...|
depicts each MO as doubly occupied (evoking the difficult imagery
of electron pairs stretching from pole to pole), whereas the
corresponding NLMO determinantal wavefunction
(24) ΨNLMO = det|...(σCH(↑))2(σCH(↓))2...|
sensibly depicts each pair as occupying a distinct localized site. Despite
the obviously greater interpretive difficulties of
(23) compared to (24), the two wavefunctions are actually
mathematically equivalent (as follows from Fock's theorem [12]). The
energy and
other physical observables calculated from ΨNLMO must therefore
be identical to those from ΨCMO, and any perceived
"delocalization effect" arising from the unitary transformation
of NLMOs to the symmetry-adapted CMOs (22a,b) must be purely
illusory. Thus, NLMOs may be said to remove a superfluous tier of "unnecessary delocalization"
that merely distracts from the chemist's
Lewis structural picture, without physical consequences.
However, the NLMOs (unlike their parent NBOs)
also incorporate the physically
important delocalizations of each
electron pair with its chemical environment, thus becoming
"semi-localized" in form. As noted above,
the occupancy of a Lewis-type NBO is typically somewhat
less than two electrons, indicative of weak delocalization
of the electron pair into adjacent non-Lewis acceptor orbitals
(particularly, vicinal antibonds). Each NLMO ωi is
composed of its parent Lewis-type NBO Ωi together with the
weak "delocalization
tail(s)" contributed by non-Lewis-type NBOs Ω*j on neighboring
centers, thus restoring the
full double-occupancy. Mathematically,
the NLMO can be expressed as
(25) ωi = η[Ωi + Σj λi→j Ω*j] ,
(i = 1,2,...,N/2)
where η is a normalizing constant and the λi→j's are small
mixing coefficients that reflect the strength of Ωi→ Ω*j
donor-acceptor interactions. These mixing coefficients
can be accurately approximated by low-order perturbation
theory, as discussed below.
More generally, the NLMOs are a complete orthonormal set
consisting of both Lewis ωi's (25) and non-Lewis ω*j's (26)
the former being all doubly occupied (<ωi|Γ|ωi> = 2) and
the latter completely vacant (<ω*j|Γ|ω*j> = 0) in
uncorrelated single-configuration MO wavefunctions. As described
below, the Lewis and non-Lewis NLMOs are actually obtained
simultaneously (and symmetrically) by a procedure that applies
equally to uncorrelated and correlated densities. In the latter
case the degenerate occupancy pattern is broken, with
some non-Lewis ω*j's acquiring slight non-zero
occupancies and some Lewis ωi's falling below full
double occupancy. However, the qualitative dominance of the N/2
Lewis-type NLMOs (25) persists in both uncorrelated and correlated
wavefunctions, reflecting the dramatic condensation of
electron density into a small subset of the full number
of NLMOs needed to span the basis space. Correlated non-Lewis NLMOs
are automatically ordered in importance by their non-zero occupancies,
facilitating efficient description of electron correlation effects
in the most compact orbital subspace. However, the role of NBOs and NLMOs
as building blocks for constructing accurate correlated
wavefunctions [13] lies beyond the
scope of present discussion.
The NLMOs are only one of several alternative sets of "localized
molecular orbitals" (LMOs) that have been suggested in the
literature. Familiar examples of the latter include
the Edmiston-Ruedenberg ERLMOs [14],
which minimize interelectron repulsions, and the Boys BLMOs [15],
which maximize the separation of orbital centroids. The final forms
of the NLMOs generally agree well with these alternative LMOs. However,
the numerical procedure for determining NLMOs differs significantly,
both in general computational strategy
(minimal delocalization of starting NBOs vs.
localization of starting CMOs) and specific criterion of localization
(maximum resemblance
to parent NBO vs. minimized repulsions or maximized centroid separations
to other LMOs). For both reasons, NLMO determination is numerically
efficient compared to alternative LMO algorithms.
Let us now briefly summarize some details of
numerical NLMO determination. The procedure starts from the NBO
density matrix bΓ (the matrix representation of Γ
in the full basis of NBOs), which is already of
near-diagonal form. The general strategy is to
zero all off-diagonal elements between Lewis (L) and non-Lewis (NL)
blocks of this matrix by a sequence of unitary 2 x 2
Jacobi rotations [16]. Each step consists of choosing
the largest remaining off-diagonal element Γi,j
(where index i is in the L-block and j in the NL-block)
and the associated rotation angle θi,j to unitarily
transform the 2 x 2 matrix with diagonal
elements Γi,i, Γj,j to diagonal form, thereby
increasing the diagonal occupancy
in the L-block and reducing it in the NL-block. For sufficiently
small off-diagonal element, the Jacobi rotation angle θi,j
can be approximately related to the mixing coefficients in (25), viz.,
(27) λi→j ≅ sin(θi,j) ≅ θi,j
(Because the initial off-diagonal NBO elements
<ωi|Γ|ω*j> are intrinsically small,
the NLMO procedure converges rapidly compared to analogous
2 x 2 Jacobi rotations for ERLMOs or BLMOs.) The final product
of Jacobi rotations block-diagonalizes the density
operator, so that L and NL blocks become non-interacting
in the NLMO limit. By construction, the L-block
NLMOs achieve maximum occupation (actually, all doubly occupied in the
single-configuration MO limit) while the NL-block NLMOs
achieve maximum depletion
(all strictly vacant in the MO limit). For further details see
the original paper [17].
The mixing coefficients to obtain semi-localized NLMOs from
strictly localized NBOs
can also be estimated by low-order perturbation
theory. We start from a matrix representation of the Fock or Kohn-Sham
(effective 1-electron Hamiltonian) operator F in the basis of
NBOs Ωi, Ω*j, the "unperturbed eigenfunctions" for the diagonal
matrix ("unperturbed Hamiltonian") with orbital energies
(28) εi = <Ωi|F|Ωi>, εj
= <Ω*j|F|Ω*j>
The off-diagonal
element Fi,j = <Ωi|F|Ω*j> provides the "perturbation" to
convert unperturbed eigenfunctions (NBOs) to final eigenfunctions
(NLMOs) of the 2 x 2 F matrix. According
to Rayleigh-Schrödinger perturbation theory [18],
the first-order approximation for eigenfunction ωi is
(29) ωi = Ωi + [Fi,j/(εi − εj)]Ω*j
while the corresponding second-order energy lowering (for 2-electron
occupancy) is
(30) ΔE(2)i→j = −2 Fi,j2/(εj − εi)
From comparison with (29), the mixing coefficient λi→j in (25) is
estimated as
(31) λi→j = Fi,j/(εi − εj)
and the occupancy transfer ("charge transfer") Qi→j
from Ωi to Ω*j is
(32) Qi→j = 2λi→j2
The perturbative expressions (29)-(32) thereby provide useful estimates
of the specific Ωi→Ω*j delocalization effects
on the energetics and composition of NLMO formation, including
the Jacobi angles in (27). Standard $NBO keywords deliver
the matrix elements εi, εj, Fi,j needed to evaluate
these expressions numerically. The 2nd-order delocalization
energies (30) for all significant donor-acceptor interactions
are a standard feature of NBO program output.
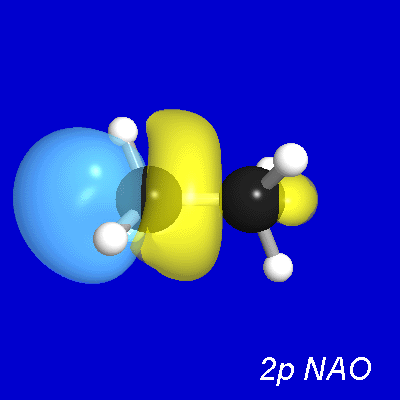
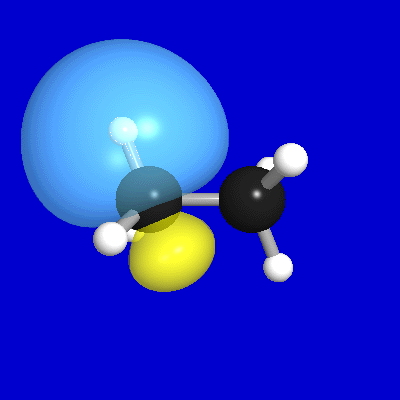
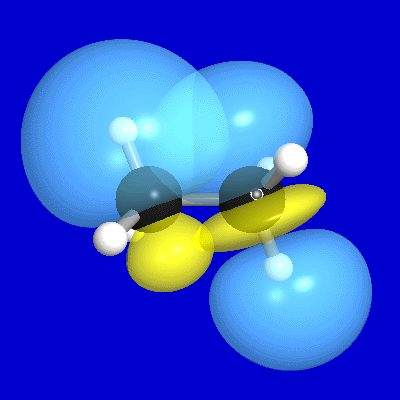
Figure 7 illustrates the leading donor-acceptor delocalization
in ethane between a donor CH bond (σCH) at C(1) and the antiperiplanar
vicinal acceptor CH antibond (σ*C'H') at C(2). As shown in the
upper panels, the vicinal σCH, σ*C'H' NBOs overlap (and interact)
significantly, primarily due to σ*C'H' "backside"
overlap with the σCH bond "shoulder." [Of course, a
complementary σC'H'→σ*CH delocalization
exists between the donor σC'H' on C(2)
and the acceptor σ*CH on C(1).] This interaction leads to
the slightly distorted forms of both the L-type NLMO
that originates from σCH (middle
panels) and the NL-type NLMO that originates from σ*C'H' (lower
panels), expressed numerically as [cf. (29)]
(33a) ωCH = 0.998σCH + 0.057σ*C'H' + ...
(33b) ω*C'H' = 0.998σ*C'H' − 0.057σCH + ...
The energy lowering for each
σCH→σ*C'H' interaction [cf. (30)]
is 2.59 kcal/mol, corresponding to weak charge delocalization [cf. (32)]
of 0.0081e in staggered geometry. Because these "hyperconjugative"
σCH→σ*C'H' delocalizations
are acutely sensitive to H-C-C'-H' dihedral angle
(vanishing, e.g., when donor and acceptor orbitals are in
orthogonal planes), their torsional
variations are the principal source of the famous
rotation barrier of ethane [19].
Figure 7:σCH,
σ*CH NLMOs of Ethane
REFERENCES
[1] P.-O. Löwdin, Phys. Rev. 97, 1474 (1955);
E. R. Davidson, Reduced Density Matrices in Quantum Chemistry
(Academic Press, New York, 1976).
[2] P. Jorgensen and J. Simons, J. Chem. Phys. 79, 334 (1983);
F. Weinhold and J. E. Carpenter, J. Mol. Struct. (Theochem) 164, 189 (1988).
[3] B. C. Carlson and J. M. Keller, Phys. Rev. 105, 102 (1957).
[4] A. E. Reed, R. B. Weinstock, and F. Weinhold,
J. Chem. Phys. 83, 735 (1985).
[5] R. S. Mulliken, J. Chem. Phys. 23, 1833, 1841, 2338, 2343 (1955).
[6] Quoted in R. S. Mulliken and W. C. Ermler, Diatomic Molecules:
Results of Ab Initio Calculations (Academic, New York, 1977), pp. 33-38.
[7] (a) J. K. Badenhoop and F. Weinhold, J. Chem. Phys. 107, 5406 (1997);
(b) J. K. Badenhoop and F. Weinhold, J. Chem. Phys. 107, 5422 (1997);
Int. J. Quantum Chem. 72, 269 (1999).
[8] J. P. Foster and F. Weinhold, J. Am. Chem. Soc. 102, 7211 (1980).
[9] A. E. Reed and F. Weinhold, J. Chem. Phys. 78, 4066 (1983).
[10] F. Weinhold, "Natural Bond Orbital Methods," in
Encyclopedia of Computational Chemistry,
P. v.R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollman,
H. F. Schaefer III, P. R. Schreiner (Eds.), (John Wiley & Sons, Chichester, UK,
1998), Vol. 3, pp. 1792-1811.
[11] F. Weinhold (ed.), NBO 5.0 Program Manual (Theoretical Chemistry
Institute, Univ. Wisconsin-Madison, 2001).
[12] V. Fock, Z. Physik 61, 126 (1930).
[13] A. V. Nemukhin and F. Weinhold, J. Chem. Phys. 97, 1095 (1992);
N. Flocke and R. J. Bartlett, Chem. Phys. Lett. 367, 80 (2003).
[14] C. Edmiston and K. Ruedenberg, Rev. Mod. Phys. 34, 457 (1963).
[15] J. M. Foster and S. F. Boys, Rev. Mod. Phys. 32, 300 (1960).
[16] J. H. Wilkinson, The Algebraic Eigenvalue Problem (Clarendon
Press, Oxford, 1965), p. 266.
[17] A. E. Reed and F. Weinhold, J. Chem. Phys. 83, 1736 (1985).
[18] I. N. Levine, Quantum Chemistry, 5th ed. (Prentice Hall,
Upper Saddle River, NJ, 2000), p. 246ff.
[19] T. K. Brunck and F. Weinhold, J. Am. Chem. Soc. 101, 1700 (1979);
V. Pophristic and L. Goodman, Nature 411, 565 (2001);
F. Weinhold, Angew. Chem. Int. Ed. 42, 4188 (2003).